Atomistic Insight into Cation Effects on Binding Energies in Cu-Catalyzed Carbon Dioxide Reduction
Abstract
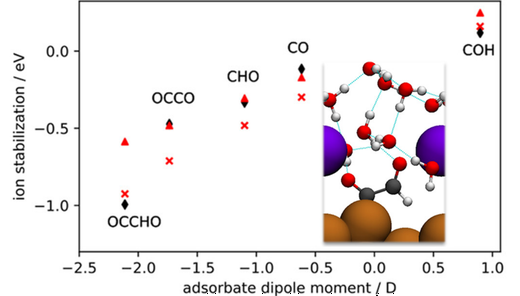
Electrolyte properties in general and electrolyte ions in particular have been shown to have significant effects on the kinetics of electrochemical CO2 and CO reduction at metal electrodes, but these effects have not yet been fully understood. We investigate the effects of cations at the aqueous electrolyte–Cu(211) interface on adsorbate binding energies and the electrolyte structure using density functional theory (DFT). Charging the interface via explicit Na+ has systematic effects on adsorbate–electrolyte interactions and conformations. We describe specific local adsorbate–ion interactions, including direct alkali ion–adsorbate coordination and hydrogen bonding via ion-coordinated water molecules. The relative importance of these specific interactions and purely electrostatic field–adsorbate interactions is investigated by comparing the DFT-calculated ion effects to those predicted by purely electrostatic models of the interface and the adsorbates. We find that the trend in ion effects among different adsorbates at constant surface charge density is well explained by a purely electrostatic interaction model. The binding energy of OCCHO is found to depend strongly on the surface charge density as well as the spatial distribution of ions at constant surface charge density. These effects are also explained by a purely electrostatic local electrostatic field–adsorbate model. This indicates that alkali ion effects can be mainly attributed to purely electrostatic field interactions and that the local field at ion-stabilized active sites can depend significantly on both the overall charge density and on the spatial distribution of ions at constant charge density. This work provides new insight on alkali ion effects on a variety of adsorbates relevant to the CO2 and CO reduction reactions by describing specific local ion–adsorbate interactions, systematic changes in adsorbate–electrolyte interactions induced by explicit surface charge, and comparisons of the calculated effects on binding energies to simple electrostatic field–adsorbate models to explain trends among various adsorbates and at varying surface charge densities and supercell configurations.