Solvent–Adsorbate Interactions and Adsorbate-Specific Solvent Structure in Carbon Dioxide Reduction on a Stepped Cu Surface
Abstract
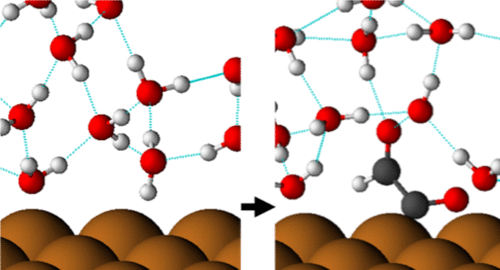
In this work, the structure of water and its interactions with various carbon dioxide reduction intermediates adsorbed on a Cu(211) surface is investigated using density functional theory. We find that the presence of adsorbates has a significant and adsorbate-specific effect on the local water structure and that solvation can stabilize adsorbate conformations different from those found in vacuum. We describe relationships between the hydrogen bonding capability of an adsorbate, the dipole moment of the adsorbate, the energetic strength of water–adsorbate interactions, and the change induced in the local water orientation by the adsorbate. Mechanistic implications are discussed. We investigate and quantify the error associated with using arbitrary locally optimized solvent structures in calculations of relevant physical quantities, such as solvated binding energies and work functions. Possible effects of thermal motion on calculations of the work function are investigated using ab initio molecular dynamics.