Using pH Dependence to Understand Mechanisms in Electrochemical CO Reduction
Abstract
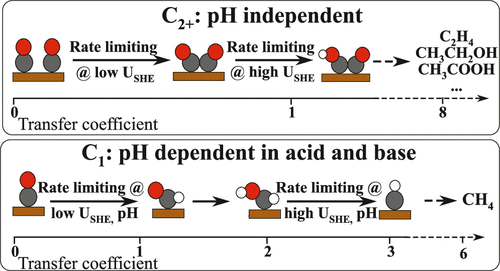
Electrochemical conversion of CO(2) into hydrocarbons and oxygenates is envisioned as a promising path toward closing the carbon cycle in modern technology. To date, however, the reaction mechanisms toward the plethora of products are disputed, complicating the search for alternative catalyst materials. To conclusively identify the rate-limiting steps in CO reduction on Cu, we analyzed the mechanisms on the basis of constant-potential density functional theory (DFT) kinetics and experiments at a wide range of pH values (3–13). We find that *CO dimerization is energetically favored as the rate-limiting step toward multicarbon products. This finding is consistent with our experiments, where the reaction rate is nearly unchanged on a standard hydrogen electrode (SHE) potential scale, even under acidic conditions. For methane, both theory and experiments indicate a change in the rate-limiting step with electrolyte pH from the first protonation step under acidic/neutral conditions to a later one under alkaline conditions. We also show, through a detailed analysis of the microkinetics, that a surface combination of *CO and *H is inconsistent with the measured current densities and Tafel slopes. Finally, we discuss the implications of our understanding for future mechanistic studies and catalyst design.