Our new paper is out in Nature Catalysis! A huge congratulations to Seungchang Han!
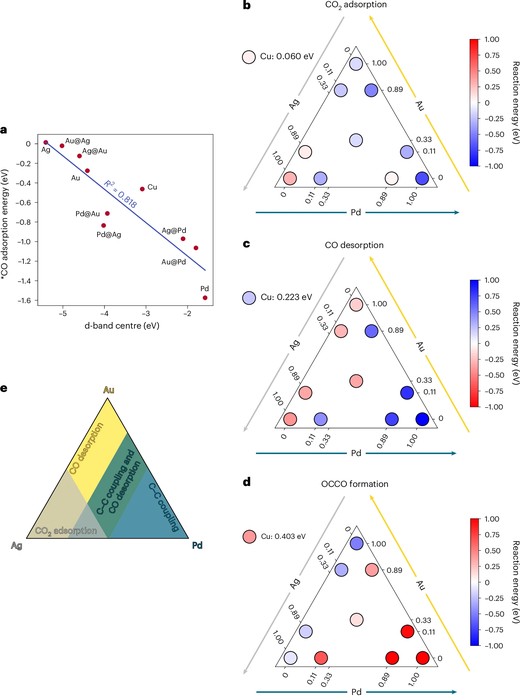
Two years ago, I proposed theoretical descriptors for CO2RR product selectivity—specifically the potential of zero charge (work function) and CO adsorption energy based on electric double layer aware DFT calculations of bare transition metals (https://lnkd.in/gWqFyKVt). The question that remained was: How do they perform in actual catalyst design? This motivated the group of Jihun Oh (KAIST) to design new non-Cu metal alloys. In our collaborative work, the experimental group showed that these descriptors can successfully predict catalysts optimized for CO and formate production. But there’s a twist: none of the materials produced C2 products, even when matching Cu-like descriptor values. Further theory from my PhD student Seungchang Han revealed that surface heterogeneity introduces adsorption sites that block specific key steps toward C2 formation. Copper remains unique in its homogeneous distribution of active sites—it binds CO2 strongly, retains CO on the surface, and enables efficient CO–CO coupling. The takeaway: descriptor-based models must evolve to capture multi-site, multi-element complexity before being deployed in high-throughput and AI-driven catalyst discovery.